







ISO発行・改訂情報
ISO13485:2016移行支援
1.ISO13485:2016の発行
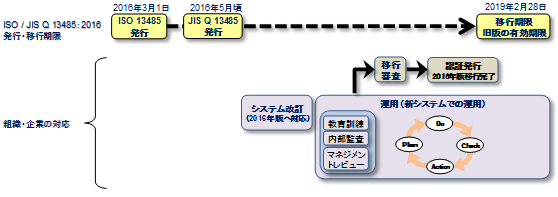
ISO13485「医療機器-品質マネジメントシステム-規制目的のための要求事項」は、第二版(2003年版)の発行から既に10年以上経過しており、改訂作業が進められていましが、ISO13485:2016は2016年3月1日に発行されました。
移行期限は、ISO13485:2016発行から3年とされていますが、移行審査から登録までに時間要することがあり、各認証機関において移行審査の期限が定められますのでご注意ください。
2.ISO13485:2016の改訂内容
今回の改訂は、主として“ISO13485固有の要求事項の強化”、“規制要求事項との整合”などを目的としています。
ISO13485:2016の構成
2016年版は、当初よりマネジメントシステムの共通構造(HLS※1)を採用しないことが決まっていたため、ISO9001:2008がベースとなっています。ISO13485:2016は、ISO13485:2003及びISO9001:2008と同じ、箇条4~8で構成されます。
※1:2015年9月に改訂されたISO9001:2015、ISO 14001:2015は、共通構造(HLS)を採用したことにより、規格の構造が大きく変わりました。HLSを採用した規格は、箇条4~10からなるPDCA(Plan:箇条4~7 Do:箇条8 Check:箇条9 Act:箇条10)で構成されています。
規格の箇条は、以下のような構成となっています。
| 4. 品質マネジメントシステム | ||
| 4.1 一般要求事項 |
||
| 4.2 文書化に関する要求事項 | ||
| 4.2.1 一般 4.2.2 品質マニュアル 4.2.3 医療機器ファイル 4.2.4 文書管理 4.2.5 記録の管理 |
||
| 5. 経営者の責任 |
||
| 5.1 経営者のコミットメント |
||
| 5.2 顧客重視 |
||
| 5.3 品質方針 |
||
| 5.4 計画 | ||
| 5.4.1 品質目標 5.4.2 品質マネジメントシステムの計画 |
||
| 5.5 責任、権限及びコミュニケーション | ||
| 5.5.1 責任及び権限 5.5.2 管理責任者 5.5.3 内部コミュニケーション |
||
| 5.6 マネジメントレビュー | ||
| 5.6.1 一般 5.6.2 マネジメントレビューへのインプット 5.6.3 マネジメントレビューからのアウトプット |
||
| 6. 資源の運用管理 | ||
| 6.1 資源の提供 |
||
| 6.2 人的資源 |
||
| 6.3 インフラストラクチャ |
||
| 6.4 作業環境及び汚染の管理 | ||
| 6.4.1 作業環境 6.4.2 汚染の管理 |
||
| 7. 製品実現 | ||
| 7.1 製品実現の計画 |
||
| 7.2 顧客関連のプロセス | ||
| 7.2.1 製品に関連する要求事項の明確化 7.2.2 製品に関連する要求事項のレビュー 7.2.3 コミュニケーション |
||
| 7.3 設計・開発 | ||
| 7.3.1 一般 7.3.2 設計・開発の計画 7.3.3 設計・開発へのインプット 7.3.4 設計・開発からのアウトプット 7.3.5 設計・開発のレビュー 7.3.6 設計・開発の検証 7.3.7 設計・開発のバリデーション 7.3.8 設計・開発の移管 7.3.9 設計・開発の変更管理 7.3.9 設計・開発のファイル |
||
| 7.4 購買 | ||
| 7.4.1 購買プロセス 7.4.2 購買情報 7.4.3 購買製品の検証 |
||
| 7.5 製造及びサービス提供 | ||
| 7.5.1 製造及びサービス提供の管理 7.5.2 製品の清浄性 7.5.3 据付け活動 7.5.4 付帯サービス活動 7.5.5 滅菌医療機器に対する特別要求事項 7.5.6 製造及びサービス提供に関するプロセスバリデーション 7.5.7 滅菌及び無菌バリアシステムのプロセスバリデーションに対する特別要求事項 7.5.8 識別 7.5.9 トレーサビリティ 7.5.9.1 一般 7.5.9.2埋込み医療機器に対する特別要求事項 7.5.10 顧客の所有物 7.5.11 製品の保存 |
||
| 7.6 監視機器及び測定機器の管理 | ||
| 8. 測定、分析及び改善 | ||
| 8.1 一般 |
||
| 8.2 監視及び測定 | ||
| 8.2.1 フィードバック 8.2.1 苦情処理 8.2.3 規制当局への報告 8.2.4 内部監査 8.2.5 プロセスの監視及び測定 8.2.6 製品の監視及び測定 |
||
| 8.3 不適合品の管理 | ||
| 8.3.1 一般 8.3.2 引き渡し前の不適合製品における処置 8.3.3 引き渡し後に発見された不適合製品における処置 8.3.4 手直し |
||
| 8.4 データの分析 | ||
| 8.5 改善 | ||
| 8.5.1 一般 8.5.2 是正処置 8.5.3 予防処置 |
||
ISO13485:2016の主な変更点(抜粋)
| ○ 適用 | ||
| ・ | 適用される組織は、2003年版に対して、より具体的に示され、“医療機器の設計・開発、製造、保管、配送、据付け、又は付帯サービス、及び設計、開発及び関連する活動(例;技術支援)の提供を含む医療機器のライフサイクル段階に関係する組織が使うことができる”とされています。 | |
| ○ 文書・記録 | ||
| ・ | 全般的に、手順等の文書化の要求が厳しくなっています。 | |
| ○ 4 品質マネジメントシステム | ||
| ・ | プロセスの管理に“リスクに基づくアプローチを考慮する”ことが求められています。 | |
| ○ 7.2 顧客関連プロセス | ||
| ・ | ユーザートレーニングに関する要求事項を明確にし、レビューすることが求められています。 | |
| ○ 7.3 設計・開発 | ||
| ・ | 製造への移管に関する手順を文書化することが求められています。 | |
| ・ | 検証・妥当性確認の計画文書が求められています。 | |
| ○ プロセスバリデーション | ||
| ・ | 「7.5.2. 製造及びサービスの提供に関するプロセスの妥当性確認」にあった“コンピュータソフトウェアの妥当性確認”が、「4 品質マネジメントシステム」でも要求されています。 さらに、プロセスバリデーションの手順を文書化することが求められています。 | |
| ○ 8.3 不適合品の管理 | ||
| ・ | 「8.3.2 引き渡し前の不適合製品における処置」、「8.3.3 引き渡し後に発見された不適合製品の管理」、「8.3.4 手直し」に分けられました。 引き渡し後の処置には、通知書の発行などが含まれます。 | |
| ○ 8 測定、分析及び改善 | ||
| ・ | 「8.2.2 苦情処理」、「8.2.3 規制当局への報告」などが箇条として追加され、運用における医療機器の実態に則したような構成となっています。 | |
3.推奨する改訂対応
品質マニュアルをISO13485:2016に合わせて改訂する。
2003版との差分理解。新規・追加事項を反映させる。
新QMSを運用して内部監査、マネジメントレビューを実施し、移行審査に備える。
4.テクノソフトのISO13485:2016移行支援
| 支援項目
|
支援内容
|
成果物/支援結果
|
|
|---|---|---|---|
A |
ISO13485:2016改訂内容の説明 |
半日程度の訪問で改訂内容について解説 | 改訂内容の理解 |
B |
システムの再構築 (品質マニュアル及び関連文書の改訂) |
文書改訂は訪問打合せで改訂後の内容を説明 | 品質マニュアル、関連文書 |
C |
教育の支援 |
改訂ISOマニュアルや関連規定に関する一般従業員向け教育資料の作成と教育実施のサポート | 教育資料 |
D |
ISO13485:2016対応 内部監査員養成セミナー |
1日の訪問セミナーで実施し修了証を交付 | ISO13485:2016対応内部監査員 |
E |
審査前準備 |
教育訓練、内部監査、マネジメントレビュー実施記録の確認と移行審査に向けての準備事項を説明 | 審査前準備の完了 |
F |
移行審査への対応 |
移行審査の指摘対応の検討と内容確認 | 移行登録証 |










